

Last Updated : February 11, 2016

Details
FilesProject Line:
Health Technology Review
Project Number:
ES0300-000
Background
Drugs for rare diseases (DRDs), also referred to as orphan drugs in some jurisdictions, are typically small-molecule drugs or biopharmaceuticals (referred to collectively herein as “drugs”) used to treat rare diseases. Due to a shift in the focus of the biopharmaceutical industry’s research and development priority from blockbuster to niche drugs, the DRD pipeline and the number of marketed DRDs are expanding.
The purpose of this Environmental Scan is to provide an overview of the DRD landscape in Canada and globally. This information may assist drug policy decision-makers, as well as other stakeholders, in understanding the landscape of DRDs and their review and reimbursement.
Several countries have legislation specific to DRDs. This legislation is designed to stimulate research into DRDs and make it financially viable for drug manufacturers through incentives. Definitions for rare diseases differ based on various jurisdictions’ related legislation or policies, and thus, there is no single definition for rare diseases that is accepted worldwide. These definitions typically include a criterion of disease incidence or prevalence; for example (see also Table 1):
- According to the Orphan Drug Act, the US FDA considers a rare disease to be one that affects fewer than 200,000 Americans.1
- Orphan drug designation by the European Medicines Agency (EMA) uses a condition prevalence in the European Union (EU) of not greater than five in 10,000 people affected (i.e., one in 2,000).2,3
- In Japan (Ministry of Health, Labour and Welfare), granting of orphan drug status is considered for drugs that meet specific criteria, including a disease affecting fewer than 50,000 people in Japan.4-6
- Australia’s Therapeutic Goods Administration, for purposes of orphan drug designation, states that a rare disease is one with a prevalence of fewer than 2,000 people in Australia at any time if the application is for a vaccine or an in vivo diagnostic agent. For medicines, the definition of prevalence or incidence of rare disease is not stated.7,8
- In South Korea (Ministry of Food and Drug Safety), rare diseases are defined as diseases affecting fewer than 20,000 people.9
- Taiwan’s Department of Health classifies a rare disease as one that is prevalent in fewer than one in 10,000 people of the population, is difficult to diagnose and treat, and has a genetic origin.9
- For Alberta’s publicly funded drug plan, a rare disease is defined as a genetic lysosomal storage disorder that occurs at a frequency of fewer than one per 50,000 Canadians.10
- Ontario’s publicly funded drug plan’s working definition of rare disease includes those with an incidence rate of fewer than one in 150,000 live births or new diagnoses per year.11
- Health Canada’s draft definition of a rare disease is one that affects fewer than five in 10,000 persons in Canada.12
Generally, rare diseases are considered to be severe, progressive, degenerative, life-threatening, or chronically debilitating, with a low prevalence.13,14 A large proportion (65% to 75%) of rare diseases have their onset in childhood.15,16 The majority (80%) of rare diseases are genetic in origin, affecting between 3% and 4% of births,13,17 or have a genetic component.14 Allergic, infectious (bacterial or viral), degenerative, proliferative and environmental (e.g., chemicals, radiation), or combinations of genetic and environmental causes have been recognized as well, yet in other cases specific causes remain unknown.14,17,18 A large percentage of rare diseases undergoing active research are cancers;18 however, DRDs encompass all therapeutic areas.
Table 1: Rare Disease Definitions
| Organization | Rare Disease Definition |
|---|---|
| US Food and Drug Administration | “The number of people affected by the disease or condition for which the drug is to be developed is fewer than 200,000 persons; or there is no reasonable expectation that the sales of the drug will be sufficient to offset the costs of developing the drug for the US market and the costs of making the drug available in the United States.”1 |
| European Medicines Agency | “The medicine must be intended for the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating; the prevalence of the condition in the EU must not be more than 5 in 10,000 or it must be unlikely that marketing of the medicine would generate sufficient returns to justify the investment needed for its development; no satisfactory method of diagnosis, prevention or treatment of the condition concerned can be authorised, or, if such a method exists, the medicine must be of significant benefit to those affected by the condition.”2 |
| Japan’s Ministry of Health, Labour and Welfare | “The number of patients who may use the drug should be less than 50,000 in Japan. The drugs should be indicated for the treatment of serious diseases, including difficult-to-treat diseases. In addition, they must be drugs for which there are high medical needs satisfying one of the following criteria: 1. There is no appropriate alternative drug or treatment. 2. High efficacy or safety is expected compared with existing products.”6 |
| Australia’s Therapeutic Goods Administration | “The medicine must be intended to treat, prevent or diagnose a rare disease; or must not be commercially viable to supply to treat, prevent or diagnose another disease or condition. For a vaccine or in vivo diagnostic agent, the application must also state that the vaccine or agent will be administered in Australia to not more than 2,000 people in each year after it is registered for use for the disease or condition.”7 |
| Health Canada (proposed definition)a | “A drug intended for the diagnosis, treatment, mitigation or prevention of a life-threatening, seriously debilitating, or serious and chronic disease or condition affecting not more than five in ten thousand persons in Canada, and the drug is not currently authorized by the Minister or if currently authorized, it will provide a potentially substantial benefit for the patient distinguishable from the existing therapy.”12 |
a The Health Canada framework is still in draft and a rare disease definition has not yet been finalized nor approved.
Although each rare disease on its own may affect only a small number of individuals, it is estimated that there are between 6,000 and 8,000 distinct rare diseases worldwide,15 with almost weekly reporting of newly identified disorders,14 and approximately 250 new diseases identified annually.9 Prevalence of rare diseases can vary among the populations of different countries.18 According to the Canadian Organization for Rare Diseases (CORD), rare diseases affect one in 12, or approximately 2.8 million Canadians.16 Thus, there is a “paradox of rarity”13 in that although individually the diseases are rare, a significant portion of a country’s population can be affected. In addition, others may be at risk for or have a rare disease, but remain unaware or undiagnosed.19 Thus, collectively, rare diseases represent a substantial health burden in Canada and worldwide, with an estimated 350 million people affected globally.20
Given the nature of rare diseases, they can significantly affect a patient’s autonomy and quality of life.21,22 For the majority of these diseases, there are currently no specific treatments available to cure or modify the disease.14 Furthermore, drug therapies that have been developed for rare diseases are usually expensive (Appendix 1). As noted by Gupta: “Above and beyond worrying about expensive therapies, patients diagnosed with a rare disease are often surprised to learn that there is limited scientific knowledge about the causes and natural history of their condition and little or no ongoing research.”23
With or without available treatments, patients face significant challenges in obtaining appropriate care, due to factors such as a lack of physician knowledge about rare diseases or symptoms of a rare disease being masked by or confused with other conditions, both of which can lead to significant delays in arriving at an accurate diagnosis.24 According to patients with rare diseases surveyed for a recent report,22 it takes US patients an average of 7.6 years and United Kingdom (UK) patients an average of 5.6 years to receive an accurate diagnosis, typically involving as many as eight physicians (four primary care and four specialists). In addition, two to three misdiagnoses are typical before arriving at a final diagnosis. Once patients are correctly diagnosed, they often face inequities and challenges in accessing any existing, approved, and typically costly, treatments. Thus patients, their families, or caregivers can be heavily burdened financially and emotionally as they seek accurate diagnosis, information, support, and treatments for these rare conditions.
Objectives
This Environmental Scan will address the following questions:
A. Drugs for Rare Diseases Regulatory Trends for FDA, European Medicines Agency, and Health Canada
- What has been the trend for orphan drug designation and approvals in the US since the enactment of the 1983 US FDA Orphan Drug Act?
- What has been the trend for orphan drug designation and approvals by the EMA since the Regulation on Orphan Medicinal Products came into force in 2000?
- What is the status of an orphan drug regulatory framework in Canada?
B. Biopharmaceutical Industry Pipeline for Drugs for Rare Diseases
- How is the DRD pipeline evolving?
- What is the current and predicted market volume of DRDs, and their financial effect?
C. Health Technology Assessment Reimbursement Frameworks for Drugs for Rare Diseases
- How are HTA and reimbursement perspectives evolving in the DRD evaluation area? (I.e., are DRD-specific evaluation frameworks used, and is cost-effectiveness a mandatory consideration?)
- Do any of Canada’s publicly funded drug plans use a DRD-specific evaluation framework to evaluate DRD funding?
Findings
A. Drugs for Rare Diseases Regulatory Trends for US FDA, European Medicines Agency, and Health Canada
Under normal drug marketing conditions, the pharmaceutical industry historically lacked incentive to commit the high costs associated with developing a new drug, estimated to be in the millions or billions of dollars per drug,25-27 to the small number of patients suffering from a specific rare disease. Expected product sales would be inadequate to cover the costs of bringing the drug to the market, making such development financially unviable for industry.28
However, over the past 30 years, research, development, and availability of innovative drugs to treat rare diseases have been enhanced through the introduction of orphan drug legislation and associated orphan drug policy economic incentives.29 Since the first orphan drug legislation was introduced in 1983 in the US (the Orphan Drug Act), other countries — including Singapore (1991), Japan (1993), Australia (1997), the EU (1999), Taiwan (2000), and South Korea (2003) — have also enacted regulatory frameworks for developing orphan drugs.9,23,30 Examples of incentives for industry to research and develop drugs to treat rare diseases vary per country and include research protocol assistance, tax incentives, expedited regulatory reviews, reducing or waiving drug application fees, tax credits, and defined periods of market exclusivity.23,31,32 (See Appendices 2 and 3.)
Orphan Drug Designation and Approval Trends in the US
The US Orphan Drug Act was enacted in 1983. Under the Act, a drug may be granted special status to treat a rare disease. For a drug to qualify for orphan designation, both the drug and the disease must meet criteria specified by the Act (see Table 1). The orphan designation qualifies the manufacturer for various development incentives (specified in Appendix 2). The regulatory requirements and process for obtaining marketing approval are the same whether or not an orphan drug designation is granted. The drug must still prove its safety and effectiveness through adequate studies.33
The US FDA has mandated the Office of Orphan Products Development to evaluate scientific and clinical data submissions from manufacturers to identify and designate DRDs. Another function of this Office is to work with the medical and research communities, professional organizations, academia, governmental agencies, industry, and rare disease patient groups on rare diseases issues. The Office also manages the Orphan Products Grants Program, which provides funding for clinical research that evaluates DRDs.34
The orphan drug legislation in the US has been highly successful in incentivizing the development of orphan drugs.35 In the eight to 10 years before its enactment, the FDA had approved only 10 DRDs for marketing.36 According to the US FDA Orphan Drug Designations and Approvals database, between 1983 and January 18, 2016,37 a total of 3,624 orphan drug designations have been made (note that this figure includes designations that have been withdrawn) and 515 of these designated drugs had received marketing approval (45 of these drugs were marketed under the Orphan Products Development Grants program).38 Based on these figures, approximately 14% of US orphan drug designations have been approved. Figure 1 summarizes the number of orphan drug designations and unique orphan drug approvals from 1984 to May 2013.
While the US Orphan Drug Act is specific to DRDs, other Regulatory Designations may be granted for drugs that do not qualify as a DRD if they meet specified criteria. These definitions are provided in Appendix 4, for information.
Figure 1: Orphan Drug Designations and Unique Orphan Drug Approvals in the US 1984 to May 2013
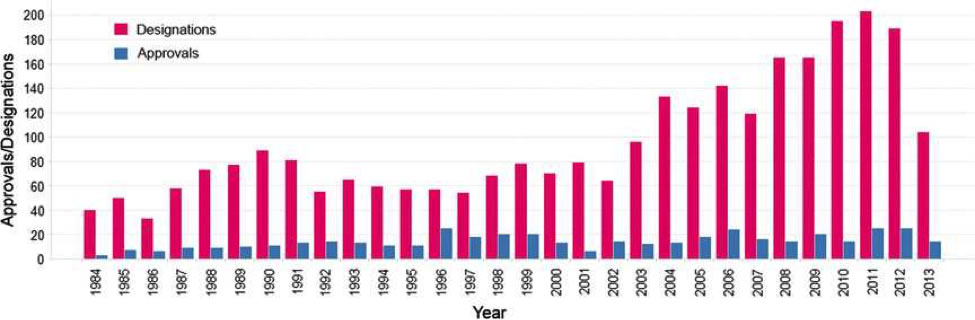
Reprinted by permission from the Royal Society of Chemistry.39
From an average of 63 designations per year in the 1990s, the number of US orphan drug designations has doubled to an average of 126 designations per year between 2001 and 2010, reflecting increased biopharmaceutical industry interest in developing drugs to treat rare diseases. The number of orphan drug approvals between 1984 and 2010 has remained relatively constant; however, as the total number of US drug approvals has been declining since the peak in the 1990s, the proportion of new drug approvals attributed to orphan drugs has risen from 17% in the 1990s to greater than 35% between 2008 and 2010.40
Figure 2 provides a breakdown of US orphan drug approvals by therapeutic area from 2006 to 2011. The majority of approvals (33%) were for oncology products; the next largest therapeutic categories, accounting for 17% of approvals each, were gastrointestinal disorders and inborn errors of metabolism, and neurological conditions. Immunological diseases and rheumatology disorders each represented 8% of approvals for this time period. The remaining 17% of approvals were other therapeutic areas, each representing less than 5%.40
Orphan Drug Designation and Approval Trends in the European Union
In the year 2000, the Regulation on Orphan Medicinal Products of the EU (Regulation [EC] No 141/2000 of December 16, 1999) came into force. As with the US Orphan Drug Act, its intention was to offer incentives to industry for developing and marketing drugs to diagnose, treat, or prevent rare conditions (Appendix 2).41
The number of orphan drug designations granted by the European Commission for the EU has increased steadily over the first 12 years of the orphan drug program. From 14 designations granted in the initial year to a total of 148 granted designations in 2012, almost 1,100 designations (not excluding expiries or withdrawals) have been granted from 2000 to 2012 inclusive.42 These orphan drug designations correspond to a total of 79 individual drugs that have received centralized marketing authorization for the EU from 2000 to 2012, representing approximately 7% of all orphan drug designations42 (Figure 3). Note that, following EMA central authorizations, individual EU member states are responsible for the funding of orphan drugs in their various jurisdictions.
Figure 2: US Orphan Drug Approvals by Therapeutic Area (2006 to 2011)
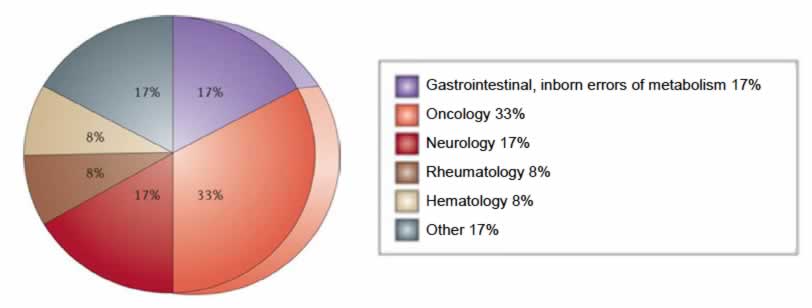
Reprinted by permission from Macmillan Publishers Ltd.43
Figure 3: European Union Orphan Drug Designations Granted and Approved; Individual Drugs Approved for Marketing (2000 to 2012)
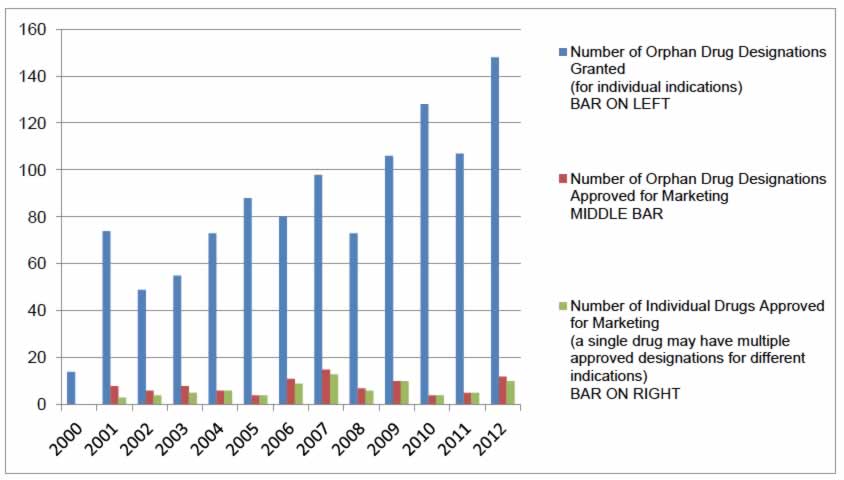
Source: Based on data retrieved from European Commission Register of Designated Orphan Medicinal Products on April 16, 2013.42
For the years 2000 to 2011, the most common therapeutic area for positive orphan drug designation opinions from the EMA’s Committee for Orphan Medicinal Products was oncology (41%), followed in decreasing order by musculoskeletal and nervous system (12%), metabolism (12%), cardiovascular and respiratory (8%), immunological (7%), anti-infectious (4%), and hematology drugs (3%). Therapeutic categories individually representing less than 3% of the total are captured under the category “other.”41
For the years 2000 to 2014, Figure 4 illustrates the distribution of opinions from the EMA by therapeutic area, for 1,430 positive opinions.44
Figure 4: European Union Positive Opinions by Therapeutic Area (2000 to 2014)
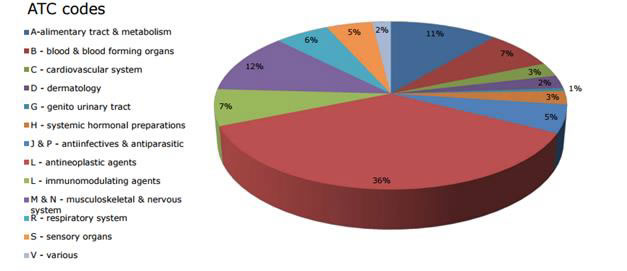
ATC = Anatomical Therapeutic Chemical.
Source: Orphan medicines figures 2000 to 2014. Updated January 1, 2015.44
Proposed Orphan Drug Regulatory Framework for Canada
Canada has no established national policies or legislative framework specifically for DRD development and market approval. On October 3, 2012, the Federal government announced the development of an orphan drug framework for Canada.45 This legislative framework would be life cycle–based, encompassing the designation, authorization, and post-market monitoring of orphan drugs, as well as incentivizing related innovative research in Canada.46 These regulations would be aligned with those of the FDA and EMA as much as possible, and are intended to provide timelier access to orphan drugs to Canadians with rare diseases.47
Health Canada has since developed a draft discussion document proposing a comprehensive orphan drug framework with a goal of providing Canadians with access to orphan drugs without compromising their safety.47 The document outlines an internationally aligned orphan drug regulatory scheme that aims to provide transparency in gathering and sharing orphan drug information with all stakeholders (including patients, health care professionals, researchers, and payers), as well as international regulatory partners.12
The Health Canada draft discussion document for the orphan drug regulatory framework12 proposes defining an orphan drug as one that meets the following criteria:
- A drug “intended for the diagnosis, treatment, mitigation or prevention of a life-threatening, seriously debilitating, or serious and chronic disease or condition affecting not more than five in ten thousand persons in Canada”, and
- A drug that “is not currently authorized by the Minister or if currently authorized, it will provide a potentially substantial benefit for the patient distinguishable from the existing therapy.”12
In keeping with orphan drug regulatory frameworks in other countries, sponsors would be asked to submit applications for orphan drug designation to Health Canada. Those meeting the application requirements, which include Health Canada’s definition of an orphan drug, would qualify for the formal granting of an orphan drug designation. In addition, Health Canada also proposes that it would recognize orphan drug designations made by other recognized international regulatory agencies (e.g., the US FDA, EU EMA).
A Health Canada orphan drug designation would subsequently provide drug sponsors with access to a number of proposed regulatory incentives, including:
- Scientific and clinical trial protocol advice by Health Canada or in common with international regulators
- Priority review of the drug submission
- Regulatory fee reductions (for small to medium enterprises)
- Linkage with the existing eight-year market exclusivity post approval (plus an additional six months for drugs with qualifying pediatric study results).
In order to promote orphan drug research, innovation, and increased potential for drugs to successfully reach the market, Health Canada’s proposed orphan drug regulatory framework would be aligned with other jurisdictions in that it would allow:
- Granting of orphan drug designations to multiple sponsors submitting applications for the same drug indicated for the same rare disease
- Granting of multiple orphan drug designations for different rare diseases to a single drug.
Because of the large number of identified rare diseases, Health Canada recognizes that it will not always have the necessary in-house scientific and medical expertise to evaluate all DRD regulatory submissions. Therefore, when necessary, the framework indicates that Health Canada will seek advice from external experts to assist in making the best possible regulatory decisions for these drugs.
In addition, patient input will be sought to provide insight into disease severity and level of unmet medical need for a particular rare disease, to be taken into consideration in the regulatory approval process. On August 6, 2014, Health Canada announced a pilot project that aims to target patient input from Canadians with rare diseases to help inform future reviews of orphan drugs, and that will simulate how patient input will be gathered and incorporated into the drug submission review process once the orphan drug framework takes effect. Patient feedback will include comments on how the rare disease affects their ability to manage their day-to-day lives, what treatments (if any) are currently available, what therapeutic benefits are most important to them, and their risk tolerance for new treatments.48
When the research for this Environmental Scan was gathered, the framework for Canada had not yet been finalized.
B. Biopharmaceutical Industry Pipeline for Drugs for Rare Diseases
1) Drugs for Rare Diseases Pipeline
The DRD pipeline is rapidly evolving. Pipeline information is, for the most part, proprietary and as such cannot readily be obtained. There are several US and European websites that may be of interest to the reader, as listed in Table 2. These websites are not specific to pipeline drugs; however, they provide an overview of the orphan drugs currently approved in the US or Europe, or that have orphan drug designation.
Table 2: Relevant DRD Websites
| Organization | Type of Information | Website |
|---|---|---|
| European Commission | Register of designated Orphan Medicinal Products | http://ec.europa.eu/health/documents/community-register/html/orphreg.htm |
| European Medicines Agency | Rare disease designations | http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/orphan_search.jsp&mid=WC0b01ac058001d12b |
| US FDA | Orphan drug designations and approvals | http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm |
| US National Institutes of Health | Rare diseases with FDA-approved medical products | https://rarediseases.info.nih.gov/gard/diseases-with-medical-products/A |
| US pharmaceutical research companies | A report on orphan drugs in the pipeline | http://www.phrma.org/sites/default/files/pdf/Rare_Diseases_2013.pdf |
DRD = drugs for rare diseases.
2) Current and Predicted Drugs for Rare Diseases Market Volume and Financial Impact
Global Orphan Drug Sales
The worldwide orphan drug market is expected to grow significantly in the future (Figure 5). A 2014 report forecasts that by 2020, global orphan drug sales will grow to $176 billion, and orphan drugs will represent 19% of the total share of prescription drug sales (excluding generics).49
Figure 5: Worldwide Orphan Drug Sales and Share of Prescription Drug Market (2000 to 2020)
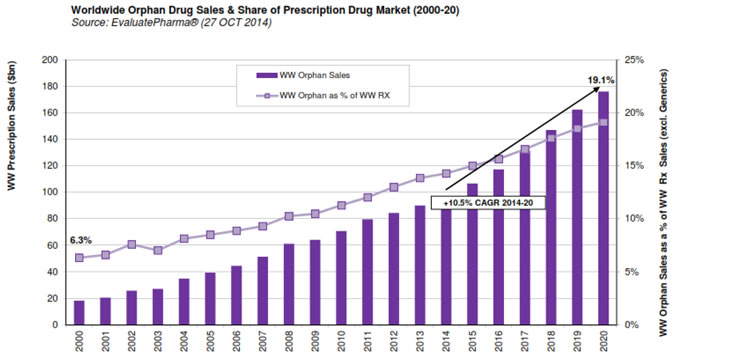
Source: EvaluatePharma Orphan Drug Report 2014, Evaluate Ltd., www.evaluate.com.50
According to the Pharmaceutical Research and Manufacturers of America (PhRMA),51 the organization that represents US-based biopharmaceutical researchers and biotechnology companies, the number of DRDs in development in the US has been rising steadily over the years. Based on PhRMA reports on the topic of rare disease, the number of drugs in development for rare diseases has tripled, from 133 drugs in 1989 to 452 in 2013 (Table 3).
Table 3: Number of Drugs for Rare Diseases in Development
| Year of PhRMA Report | Number of Drugs in Clinical Trials or Awaiting FDA Review |
|---|---|
| 1989 | 133 |
| 1991 | 176 |
| 2007 | 303 |
| 2011 | 460 |
| 2013 | 452 |
PhRMA = Pharmaceutical Research and Manufacturers of America.
Source: Table based on information from PhRMA.51,52
In 2013, the majority of DRDs being developed in the US were for the treatment of rare cancers (185 drugs). Genetic disorders (85 drugs), neurological disorders (32 drugs), and infectious diseases (28 drugs) represented the next three largest therapeutic categories specifically identified.52
C. Health Technology Assessment Reimbursement Frameworks for Drugs for Rare Diseases
1) Health Technology Assessment and Reimbursement Perspectives
A 2015 research paper reported that among the top 20 gross domestic product countries, most utilize a centralized review process as the first step in reviewing DRDs, and some utilize a complementary type of program to reconsider drugs that failed to receive a positive recommendation.53 The report notes that none of the reviewed countries has created a separate review process for DRDs, with the exception of the UK.53
Table 4 summarizes how DRDs are reviewed for the following selected HTA organizations:
- Canada's Drug Agency (the Canadian Agency for Drugs and Technologies in Health) in Canada
- Australia’s Pharmaceutical Benefits Advisory Committee (PBAC)
- Haute Autorité de Santé (HAS) in France
- Germany’s Institute for Quality and Efficiency in Health Care (IQWiG)
- Scotland’s Scottish Medicines Consortium (SMC)
- National Institute for Health and Care Excellence (NICE) in the UK.
Table 4: HTA Reimbursement Perspectives for Selected HTA Organizations
| Country (HTA Organization) | Separate Reimbursement Review Process for DRDs (Yes/No)? | Details of Process, When Applicable | Is Cost-Effectiveness a Required Element of the Reimbursement Submission? |
|---|---|---|---|
| Canada (CADTH) | No; regular submission and review process through CDR or pCODR is currently followed. | Not applicable | Yes |
| Australia (PBAC, LSDP) |
No specific DRD evaluation program for Australia, but potential options for funding DRDs through LSDP, following PBAC acceptance as clinically effective, but listing rejected due to failing required cost-effectiveness criteria. PBAC submission guidance states: “PBAC is aware of, and sympathetic to, the difficulties faced by sponsors of orphan drugs. Furthermore, the committee does not set a minimum standard for the type and level of evidence or other information that can be included in a submission to PBAC. However, it would be unlawful for PBAC not to consider comparative costs and effectiveness.”54 The “rule of rescue” may be applied as a supplement to orphan drug submissions if the 4 delineated factors are present (see below); however, the rule does not replace consideration of evidence-based comparative cost-effectiveness. PBAC has a “rule of rescue,” with the following 4 factors:54
Submissions for rare diseases that
|
LSDP Funding:55 A drug must meet the following criteria:54
There are also specific patient eligibility criteria for the drug, as well as ongoing monitoring requirements that, once approved for therapy, must be met for initial, as well as ongoing, LSDP funding. These are outlined in the various Medical Condition Guidelines Templates.56 |
PBAC (initial submission) — yes LSDP (PBAC rejected due to cost-effectiveness) — initial PBAC submission stands and only additional economic requirement for LSDP is to indicate if the drug has been previously rejected for PBS listing due to failing cost-effectiveness criteria. |
| France (HAS) | No publicly available information found regarding a separate process. | Not applicable | Not applicable (HAS does not examine any economic evidence)57 |
| Germany (IQWiG) | No publicly available information found regarding a separate process. | There is no process described per se, although the following has been stated:58 “Orphan drugs have a special status in the early benefit assessments of pharmaceuticals with new active ingredients. In accordance with statutory requirements (SGB V, section 35a, paragraph 1, sentence 10) the additional medical benefit of these medications is already proved through market authorization. Proof of medical benefit and additional medical benefit over an appropriate comparator need not be submitted. Only the extent of additional benefit must be proved for the number of patients and patient groups for whom a therapeutically significant additional benefit exists (G-BA rules of procedure, chapter 5, section 12, number 1, sentence 2). In principle, this statutory provision assumes an additional benefit for the orphan drug authorized. It does not require a relevant scientific assessment of the pharmaceutical as a foundation. Based on this statutory requirement, the G-BA determines the extent of additional benefit for orphan drugs with revenues not exceeding 50 million euros in the past 12 months based on market authorization and its substantiating studies. This limitation on the benefit assessment of orphan drugs resulting from the link to market authorization no longer applies if revenues from the pharmaceutical received through the statutory health insurance at pharmacy retail prices, including VAT, exceed 50 million euros over the past 12 months.” |
Yes |
| Scotland (SMC) | No. The SMC has changed the way it evaluates end-of-life medicines and medicines to treat very rare conditions. As of May 2014, pharmaceutical companies are able to request that SMC convenes a PACE group (see next column).59 |
Pharmaceutical companies will be asked to state in their SMC Submissions whether the medicine is in 1 of 3 categories (end-of-life medicine, orphan medicine, or ultra-orphan medicine) and to provide supporting evidence and rationale.
A submission for an end-of-life or orphan medicine will be made using the same submission form as before. The medicine will be evaluated by the NDC in the usual way. If the advice for the medicine is “not recommended” following NDC, the pharmaceutical company can choose to request that SMC convenes a PACE meeting. Each PACE group will be tailored to the medicine under consideration. The aim of the PACE group is to describe the added benefits of the medicine, from both patient and clinician perspectives, that may not be fully captured within the conventional clinical and economic assessment process, including but not limited to clinical issues, added value of the medicine for the patient, and added value of the medicine for the patient’s family and/or carers.
A submission for an ultra-orphan medicine used in extremely rare conditions will be assessed in a different way from the current process, although the submission will move through the NDC and the SMC in the same way as before. To assess ultra-orphan medicines, SMC will use a framework of explicit decision-making criteria, including the nature of the condition, impact of the medicine, impact of the technology beyond direct health benefits and on specialist services, costs to the NHS and Personal Services, and value for money. A cost-effectiveness ratio will still be requested as part of the company submission, but there may be circumstances where the choice of economic appraisal methodology has to be more flexible, given the available data and nature of the condition. It is important to capture clinicians’ and patients’ views on ultra-orphan medicines through the PACE approach, if required, and this would happen in the same way as described under end-of-life or orphan medicines. Patient Access Schemes |
Yes |
| UK (NICE)61 | Yes. Effective April 2013, NICE is responsible for coordinating the evaluation of expensive ultra-rare orphan drugs. An interim method that builds on the framework used by AGNSS has been developed for the evaluation of highly specialized drugs. The Highly Specialized Technologies Programme considers only drugs for very rare conditions, and these evaluations are recommendations on the use of new and existing highly specialized medicines and treatments within the NHS in England.62 |
The evaluation of technologies by the Highly Specialised Technologies Programme engages a specific evaluation committee that is an independent advisory body. The committee — comprising individuals who work in the National Health Service, pharmaceutical and medical devices industries, patient and caregiver organizations, and relevant academic disciplines — makes recommendations to NICE for or against the use of a technology based on its costs and benefits. “Given the very small numbers of patients living with these very rare conditions a simple utilitarian approach, in which the greatest gain for the greatest number is valued highly, is unlikely to produce guidance which would recognize the particular circumstances of these very rare conditions. These circumstances include the vulnerability of very small patient groups with limited treatment options, the nature and extent of the evidence, and the challenge for manufacturers in making a reasonable return on their research and development investment because of the very small population treated.”63 The following criteria are taken into consideration during the evaluation:
|
NICE (Highly Specialised Technologies Programme) —yes |
2) Drugs for Rare Diseases–Specific Reimbursement Programs in Canada
In Canada, following the review of new drugs through the Canada's Drug Agency Common Drug Review (CDR) or the Canada's Drug Agency pan-Canadian Oncology Drug Review (pCODR), reimbursement decisions are made at the provincial and territorial levels. All jurisdictions have general reimbursement processes, while five provinces have established processes for DRD reimbursement: British Columbia, Alberta, Saskatchewan, Ontario, and New Brunswick.53,64 In a recent publication, Menon et al. provided a detailed review of the mechanisms through which provincial and territorial drug plans provide reimbursement of drugs, including DRDs.64 The reader is referred to this work for further information.
Additional information regarding Alberta and Ontario’s DRDs programs is provided below. Of note, the New Brunswick provincial government provides coverage through the New Brunswick Drugs for Rare Diseases Plan, which assist patients with rare diseases who face high drug costs.65 Partnering with Ontario, it includes drugs that have been reviewed through the Ontario DRD Framework.65
Alberta
Alberta has a Rare Disease Drug Program for eligible Albertans under its publicly funded drug plan. This program was developed for ethical and compassionate reasons to help affected individuals with the exceptionally high costs of DRDs.66
For the purposes of this program, a rare disease is defined as a genetic lysosomal storage disorder occurring in fewer than one in 50,000 Canadians. Drug products for the treatment of the following diseases are considered for coverage: Gaucher Disease, Fabry Disease, Mucopolysaccharidosis I (MPS I) Hurler/ Hurler-Scheie, Hunter Disease, and Pompe Disease.67
Submitted applications are reviewed by Alberta’s Rare Disease Clinical Review Panel, which is a Ministry-appointed panel consisting of rare disease–treating specialists and other health care professionals with related clinical expertise.67
Rare Disease Drug Coverage Applications can be submitted for an individual patient by their “rare disease specialist,” as defined by each drug’s eligibility criteria. When applying for coverage of a DRD, applicants must consent to a number of conditions should coverage be approved. These conditions are as follows:
- Conditional initial and continued coverage are dependent upon clinical outcomes.
- Ongoing clinical outcome monitoring is mandatory.
- Inadequate patient response or deterioration, as defined by pre-established withdrawal criteria for a specific drug and/or as assessed by the program’s clinical review panel, will dictate coverage discontinuation.67
Note that the presence of a significant illness likely to affect life expectancy, outside of the rare disease itself, is considered a contraindication to the rare disease funding.67
In addition to its DRD-specific reimbursement program, the Alberta government has a Short-Term Exceptional Drug Therapy program that allows for funding consideration for certain therapies without current public or private funding options.68 This may include drugs with or without market authorization.64
Ontario
The Ontario Ministry of Health and Long-Term Care developed a separate evaluation framework for assessing funding of DRDs under its publicly funded drug program. A framework was needed due to the absence of a national strategy for reviewing and evaluating DRDs and because of a historic and ongoing need to address patient access to these drugs in Ontario.69
This evaluation is conducted by a separate five-member DRD Working Group, which has physician, economist, pharmacist, and geneticist representation and reports directly to the Executive Officer of the Ontario Public Drug Programs.70 There are no restrictions on the types of rare diseases considered for evaluation under this framework, and requests for funding consideration under the DRD framework may be submitted by manufacturers or physicians.69
The evaluation framework uses an evidence-based process and considers the best achievable evidence for the particular drug under review, with indirect evidence taken into consideration when necessary. Predictive models identifying the natural progression of a disease, where a drug might provide the treatment effect, and the patients most likely to benefit from treatment, are used to inform the funding decisions.70
The framework consists of seven steps:11,69,70
Step 1 – Assess whether a submitted disease meets the framework’s criterion of “rare”
Determination of whether a DRD is eligible for review under the framework is determined by:
- A disease incidence rate of fewer than one in 150,000 live births or new diagnoses per year
- The lack of availability or feasibility of adequately powered randomized controlled trials detecting clinically relevant outcomes, given the rarity of the disease.
Step 2 – Gain an understanding of the natural history of the disease
This step entails a review of the disease state itself, including its presentation, progression over time, underlying mechanism, and consequences, as background information for understanding the mechanism(s) of action of the drug candidate under review.
Step 3 – Assess the potential effectiveness of the drug based on the best available evidence
This step evaluates the clinical evidence supporting the drug candidate. This includes direct clinical trial data on the drug and indirect evidence (e.g., from other disease conditions) where appropriate. When only sparse or questionable clinical data are available, use of the Bradford Hill criteria for causality, adapted for treatment assessment, is applied as a tool in assessing whether the drug alone potentially caused the reported patient benefits or not.69
Step 4 – Evaluate budget and cost impact
Cost-effectiveness analyses are not conducted; cost-effectiveness is not a deciding factor in evaluating a drug under review by this framework. However, the affordability of the product is taken into consideration in formulating the final decision.
Step 5 – Identify whether any additional follow-up data are needed
This step identifies whether and what types of other studies may be required to generate more information.
Step 6 – Review the drug evaluation with disease experts and stakeholders
During this step, outcomes of the review (inputs, assumptions, and outputs) are shared with physician and patient stakeholder groups to identify areas of disagreement or error.71
Step 7 – Reassess
This step allows for review and incorporates new information regarding disease incidence and natural history, as well as effectiveness or cost of the reviewed drug.71
The following drugs have been evaluated through Ontario’s framework:
- Aldurazyme (laronidase) for the treatment of Hurler and Hurler-Scheie forms of MPS I
- Elaprase (idursulfase) for the treatment of Hunter Syndrome
- Ilaris (canakinumab) for the treatment of Cryopyrin-Associated Periodic Syndrome
- Myozyme (alglucosidase alfa) for both infantile and adult or late onset Pompe Disease
- Zavesca (miglustat) for the treatment of Niemann-Pick Type C Disease.11
Summary
This Environmental Scan provided an overview of legislation and reimbursement frameworks for DRDs from key organizations.
In countries with regulations in place to stimulate the development of drugs used to treat rare diseases, overall orphan drug designations and subsequent market approval are increasing. Enabling legislations include incentives such as a period of market exclusivity, accelerated market-approval reviews, regulatory fee reductions, scientific advice, and tax incentives. At the time of this update, Canadian regulations and policies to facilitate orphan drug development and market approval have not been finalized, although an orphan drug framework is currently being established.
Of the HTA organizations scanned for publicly available information, only NICE has a DRD-specific evaluation framework. The other agencies (CDR and pCODR in Canada, PBAC and LSDP in Australia, and SMC in Scotland) currently evaluate DRDs through their standard processes. DRD-specific reimbursement programs exist in five Canadian provinces: British Columbia, Alberta, Saskatchewan, Ontario, and New Brunswick. Ontario has an evaluation framework to support its reimbursement program.
Appendix 1: Rare Disease Drug Cost Examples
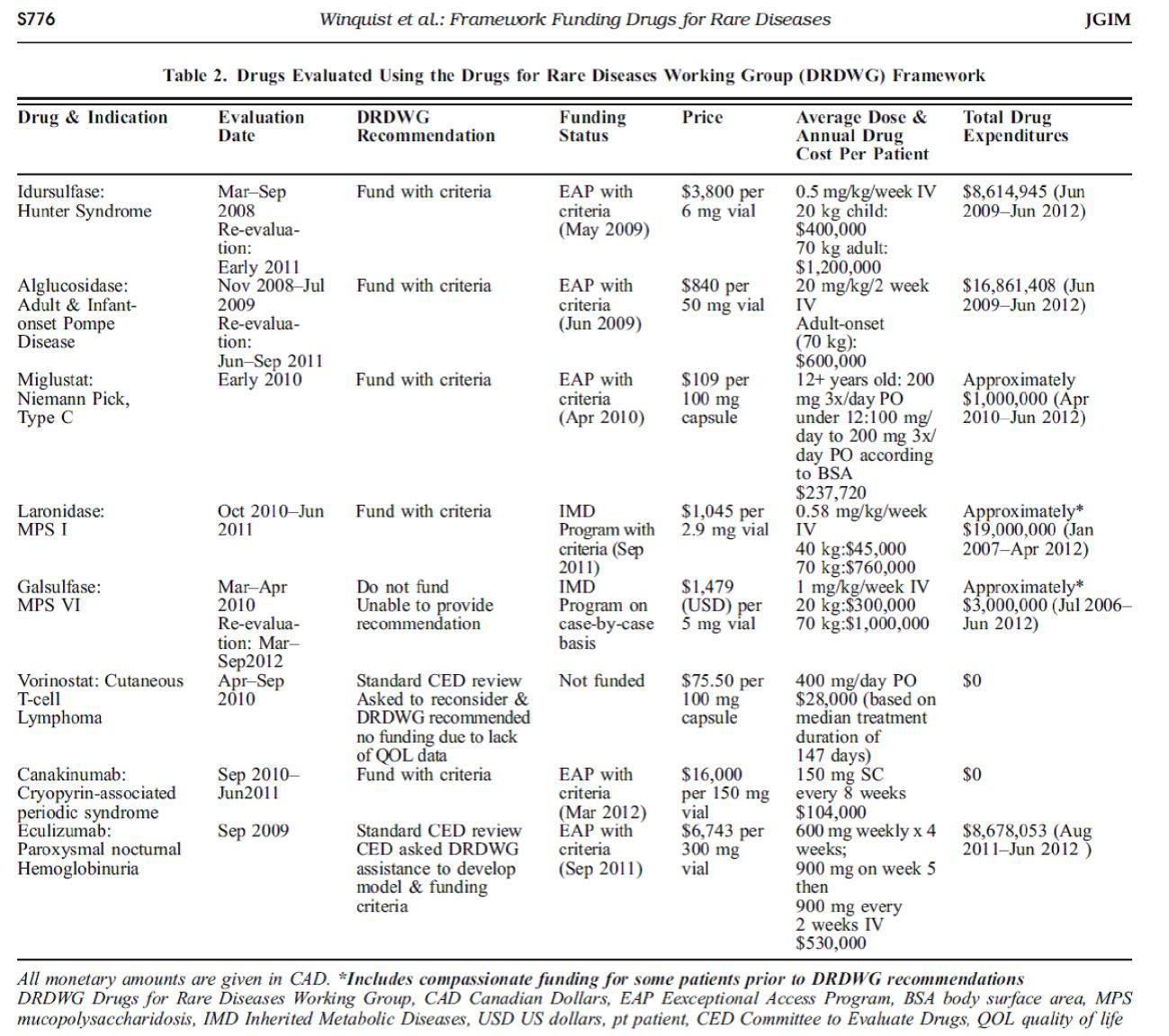
Reprinted by permission from Springer Science+Business Media.71
Appendix 2: United States and European Union Orphan Drug Enabling Legislation, Designation Criteria, and Industry Incentives
| Enabling Legislation | Criteria for Orphan Drug Designation | Incentives for Pharmaceutical Industry |
|---|---|---|
| US FDA | ||
| Orphan Drug Act 1983 (and 1984, 1985, 1988, 1992 amendments |
Regarding market exclusivity:
|
|
| EMA | ||
| The Regulation on Orphan Medicinal Products 1999 – (EC) No 141/2000 and (EC) No 847/2000 |
Regarding marketing exclusivity:
|
|
EC = European Commission; EMA = European Medicines Agency; EU = European Union; MAH = marketing authorization holder.
Appendix 3: Orphan Drug Enabling Legislation, Designation Criteria, and Industry Incentives
| Legislation and Provisions | US (FDA) | EU (EMA) | Australia (TGA) | Japan (MHLW) |
|---|---|---|---|---|
| Orphan Drug Legislation or Policy | Orphan Drug Act 1983 | Regulation on Orphan Medicinal Products 1999 | Orphan Drug Program 1997 | Orphan Drug Regulation 1993 |
| Marketing Exclusivity Period | 7 yearsa | 10 years (extended by 2 years for medicines that also comply with required pediatric investigations) | 5 years | 10 years |
| Accelerated Evaluation Availability | Yes | Yes | Yes | Yes |
| Application or Other Regulatory Fee Reductions or Waivers | Yes | Yes | Yes | No |
| Scientific Advice (Research Protocols, Technical Assistance, etc.) | Yes | Yes | Yes | Yes |
| Tax Incentives | 50% tax credits for clinical research costs | Tax credits developed by each member state | No | Tax Exemption Law (12% of expenses) |
| Other | Clinical research funding through Orphan Products Grants Program Voucher programb |
Access to EU centralized authorization process Additional incentives for micro, small, and medium-sized enterprises, including administrative and procedural assistance, and fee reductions |
Non-financial incentives include pre-licensing access and Regulatory Assistance | Extension of registration validity period Development costs partially reimbursed |
EMA = European Medicines Agency; EU = European Union; MHLW = Ministry of Health, Labour, and Welfare; TGA = Therapeutic Goods Administration.
Source: Table based on data from 4 reports.23,31,32,82
a The FDA is considering a new provision to the legislation that would add 6 months to the exclusivity period of an approved drug already on the market when the FDA approves a supplemental application for that drug for a new indication to prevent, diagnose, or treat a rare disease or condition.83
b The FDA had instituted a voucher program that awards a transferable voucher, under specified conditions, to a sponsor of an approved new drug or biological product for a rare pediatric disease. This voucher may be used for the priority review of another application. This program is currently under review.83
Appendix 4: Other Regulatory Designation Definitions
| Term | Health Canada | US FDA |
|---|---|---|
| Fast Track | NA | “Fast track is a process designed to facilitate the development, and expedite the review of drugs to treat serious conditions and fill an unmet medical need. Filling an unmet medical need is defined as providing a therapy where none exists or providing a therapy which may be potentially better than available therapy.”84 |
| Breakthrough Therapy Designation | NA | “Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint. Clinically significant endpoint generally refers to an endpoint that measures an effect on irreversible morbidity or mortality or on symptoms that represent serious consequences of the disease.”85 |
| Priority Review Designation |
“The policy applies to a submission for a serious, life-threatening or severely debilitating disease or condition for which there is substantial evidence of clinical effectiveness that the drug provides:
Priority Review status allows for a shortened review target of 180 calendar days.”86 |
“Drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. Significant improvement may be demonstrated by the following examples:
FDA’s goal is to take action on an application within 6 months.”87 |
NA = not applicable.
References
- Orphan Drug Act. Relevant excerpts (public law 97-414, as amended): Last updated January 21 2016 [Internet]. Silver Spring (MD): U.S. Food and Drug Administration; 2013 Jun 12. [cited 2016 Jan 25]. Available from: http://www.ecfr.gov/cgi-bin/text-idx?SID=bdad64762fe0b9ea486b2c9ecd6619b0&mc=true&node=se21.5.316_121&rgn=div8
- European Medicines Agency. Human regulatory [Internet]. London: The Agency. Orphan designation; 2013 May 22 [cited 2015 Dec 4]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000029.jsp&mid=WC0b01ac05800240ce
- Rodwell C, Ayme S. Rare disease policies to improve care for patients in Europe. Biochim Biophys Acta [Internet]. 2015 Oct [cited 2015 Oct 28];1852(10 Pt B):2329-35. Available from: http://www.sciencedirect.com/science/article/pii/S0925443915000599
- Orphan Druganaut Blog. Rare disease and orphan drug regulation in Japan [Internet]. In: Orphan Druganaut Blog. New York (NY): Orphan Druganaut Blog; 2013 Jan 26 [cited 2015 Dec 4]. Available from: http://orphandruganaut.wordpress.com/2013/01/26/rare-disease-and-orphan-drug-regulation-in-japan/.
- Orphanet [Internet]. version 4.13. Paris: INSERM. Orphanet: about orphan drugs; 2015 Dec 3 [cited 2015 Dec 4]. Available from: http://www.orpha.net/consor/cgi-bin/Education_AboutOrphanDrugs.php?lng=EN
- Ministry of Health, Labour and Welfare. Overview of orphan drug/medical device designation system [Internet]. Tokyo, Japan: Pharmaceutical and Food Safety Bureau; 2015. [cited 2015 Sep 28]. Available from: http://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/orphan_drug.html
- Applications for orphan drug designation [Internet].Australian Government; 2015 Jun 30. [cited 2015 Sep 17]. Available from: https://www.tga.gov.au/book/how-apply-orphan-drug-designation
- Part 3B - Orphan Drugs [Internet].Australian Government; 2015 Aug 4. [cited 2016 Jan 25]. (Therapeutic Goods Regulations 1990 - F2015C00654). Available from: https://www.comlaw.gov.au/Details/F2015C00654/Html/Text#_Toc426980968
- Song PP, Gao JJ, Inagaki Y, Kokudo N, Tang W. Rare diseases, orphan drugs, and their regulation in Asia: Current status and future perspectives. Intractable & Rare Diseases Research [Internet]. 2012 [cited 2015 Dec 4];1(1):3-9. Available from: http://www.irdrjournal.com/getabstract.php?id=511
- Alberta Human Services. Alberta Human Services drug benefit supplement [Internet]. Edmonton: Alberta Blue Cross; 2015 Apr 1. [cited 2015 Sep 17]. Available from: https://www.ab.bluecross.ca/dbl/pdfs/hsdbs.pdf
- Ontario Ministry of Health and Long-Term Care. Ontario Public Drug Programs [Internet]. Toronto: The Ministry. Drugs for rare Diseases (DRD) screening template; 2011 Aug 21 [cited 2016 Jan 25]. Available from: http://www.health.gov.on.ca/en/pro/programs/drugs/how_drugs_approv/review_rare_diseases.aspx
- Office of Legislative and Regulatory Modernization,Policy, Planning and International Affairs Directorate, Health Products and Food Branch. Initial draft discussion document for a Canadian orphan drug regulatory framework [Internet]. Ottawa: Government of Canada; 2012 Dec 13. [cited 2016 Jan 18]. Available from: http://www.orpha.net/national/data/CA-EN/www/uploads/Initial-Draft-Discussion-Document-for-A-Canadian-Orphan-Drug--Regulatory-Framework.doc
- European Organisation for Rare Diseases (EURORDIS). Rare Diseases: understanding this public health priority [Internet]. Paris: EURORDIS; 2005. [cited 2015 Dec 4]. Available from: http://www.eurordis.org/sites/default/files/publications/princeps_document-EN.pdf
- National Academy of Sciences,Institute of Medicine. Profile of rare diseases [Internet]. In: Field MJ, Boat MF, editors. Rare diseases and orphan products: accelerating research and development. Washington(DC): The Academy; 2013. p. 42. Chapter 2 [cited 2015 Dec 4]. Available from: http://books.nap.edu/openbook.php?record_id=12953&page=42.
- What is a rare disease? [Internet].EURODIS - Rare Diseases Europe; 2015. [cited 2015 Sep 17]. Available from: http://www.eurordis.org/sites/default/files/publications/Fact_Sheet_RD.pdf
- Canadian Organization for Rare Disorders (CORD): Our work [Internet]. Toronto: Canadian Organization for Rare Disorders. 2015 [cited 2015 Sep 17]. Available from: https://www.raredisorders.ca/our-work/
- European Medicines Agency [Internet]. London: The Agency. Medicines for rare diseases; 2013 [cited 2015 Dec 4]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000034.jsp&mid=WC0b01ac058002d4eb
- Azie N, Vincent J. Rare diseases: the bane of modern society and the quest for cures. Clin Pharmacol Ther. 2012 Aug;92(2):135-9.
- Canadian Organization for Rare Disorders (CORD) [Internet]. Toronto: CORD. Canadian Organization for Rare Disorders: About CORD; 2013 [cited 2015 Dec 4]. Available from: https://www.raredisorders.ca/about-cord/
- The Global Genes Project. [Internet]. Aliso Viejo(CA ): The Project. RARE diseases: Facts and statistics; 2013 [cited 2015 Dec 4]. Available from: http://globalgenes.org/rarefacts/
- Elger S. Rare disease management: experiences from abroad (summary). ETMIS [Internet]. 2011 [cited 2015 Dec 4];7(6). Available from: http://www.inesss.qc.ca/fileadmin/doc/INESSS/Rapports/OrganisationsSoins/INESSS_Summary_RareDisease_EN.pdf
- Rare Disease Impact Report: insights from patients and the medical community. Lexington(MA): Shire plc; 2013.
- Gupta S. Rare diseases: Canada's "research orphans". Open Med [Internet]. 2012 [cited 2015 Dec 4];6(1):e23. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3329116/pdf/OpenMed-06-e24.pdf
- Tang W, Makuuchi M. Intractable and rare diseases research [editorial]. Intractable & Rare Diseases Research [Internet]. 2012 [cited 2015 Dec 4];1(1). Available from: http://www.irdrjournal.com/files/IRDR_2012Vol1No1_pp1_44.pdf
- Collier R. Drug development cost estimates hard to swallow. CMAJ [Internet]. 2009 Feb 3 [cited 2015 Dec 4];180(3):279-80. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2630351/
- BIOTECanada. BIOTECanada legislative proposals to create a national orphan product policy [Internet].2008. [cited 2015 Dec 4]. Available from: http://www.biotech.ca/uploads/btc%20leg%20proposals.pdf
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ [Internet]. 2003 [cited 2015 Dec 4];22:151-85. Available from: http://moglen.law.columbia.edu/twiki/pub/LawNetSoc/BahradSokhansanjFirstPaper/22JHealthEcon151_drug_development_costs_2003.pdf
- Tambuyzer E. Rare diseases, orphan drugs and their regulation: questions and misconceptions. Nat Rev Drug Discov. 2010 Dec;9(12):921-9.
- Meekings KN, Williams CSM, Arrowsmith JE. Orphan drug development: an economically viable strategy for biopharma R&D. Drug Discov Today [Internet]. 2012 [cited 2015 Dec 4];17(13/14). Available from: http://csmres.co.uk/cs.public.upd/article-downloads/Meekings-Reuters-paper.pdf
- Orphan drugs in Asia-Pacific: from designation to pricing, funding and market access. Pharma Lett [Internet]. 2010 Feb 8 [cited 2015 Dec 4]. Available from: http://www.thepharmaletter.com/file/40975/orphan-drugs-in-asia-pacific-from-designation-to-pricing-funding-and-market-access.html
- Policy alternatives for treatments for rare diseases. CMAJ [Internet]. 2010 Nov 23 [cited 2015 Dec 4];182(17). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2988562/pdf/182e787.pdf/
- Franco P. Orphan drugs: The regulatory environment. Drug Discov Today. 2013 Feb;18(3-4):163-72.
- Designating an orphan product: drugs and biological products [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2013 Aug 14. [cited 2015 Sep 29]. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/default.htm
- Developing products for rare diseases [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2015 Oct 19. [cited 2016 Jan 25]. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/ucm2005525.htm
- Woodcock J. The future of orphan drug development. Clin Pharmacol Ther. 2012 Aug;92(2):146-8.
- Haffner ME. Adopting orphan drugs--two dozen years of treating rare diseases. N Engl J Med. 2006 Feb 2;354(5):462-71.
- Orphan drug product designation database [Internet]. Silver Spring (MD): U.S. Food and Drug Administration; 1983 - [cited 2015 Dec 4]. Available from: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm
- Information of the Orphan Products Grants Program [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2015 Oct 27. [cited 2016 Jan 25]. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/WhomtoContactaboutOrphanProductDevelopment/ucm134580.htm
- Pryde DC, Palmer MJ. Figure 1.1, Orphan drug designations and unique orphan drug approvals 1984-2013 [Internet]. In: Orphan drugs and rare diseases. 2014. p. 4 [cited 2015 Sep 30]. (RSC Drug Discovery (book 38)). Available from: http://pubs.rsc.org/en/content/chapterhtml/2014/bk9781849738064-00003?isbn=978-1-84973-806-4&sercode=bk.
- Melnikova I. Rare diseases and orphan drugs. Nat Rev Drug Discov. 2012 Apr;11(4):267-8.
- Overview of rare disease activities in Europe: part 1 [Internet]. In: Aymé S, Rodwell C, editors. 2012 report on the state of the art of rare disease activities in Europe of the European Union Committee of Experts on Rare Diseases. Paris(FR): European Union Committee of Experts on Rare Diseases(EUCERD); 2013 [cited 2015 Dec 4]. Available from: http://ec.europa.eu/health/rare_diseases/docs/eucerd2012_report_state_of_art_rare_diseases_activities_1.pdf.
- European Commission. Community register of medicinal products [Internet]. Brussels: Directorate General Health & Consumers. Register of designated Orphan Medicinal Products (by number); 2015 Dec 3 [cited 2015 Dec 4]. Available from: http://ec.europa.eu/health/documents/community-register/html/orphreg.htm
- Melnikova I. Orphan drug approvals by therapeutic area (2006-2011) [figure] [Internet]. In: Rare diseases and orphan drugs. 2012 [cited 2015 Dec 17]. (Nature Reviews Drug Discovery). Available from: http://www.nature.com/nrd/journal/v11/n4/fig_tab/nrd3654_F3.html.
- Distribution of opinions by therapeutic area from year 2000 - 2014. [Internet]. In: Orphan medicines figures 2000/2014. London: European Medicines Agency; 2015 [cited 2015 Dec 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/04/WC500185766.pdf.
- Health Canada. Press release. Ottawa: Government of Canada. Harper Government takes action to help Canadians with rare diseases: Launch of first ever Canadian framework to increase access to new treaments and information and Orphanet-Canada online portal; 2012 Oct 3
- Health Canada. Press release. Ottawa: Government of Canada. An orphan drug framework for Canada; 2013 Oct 3
- Lee DK, Wong B. An Orphan Drug Framework (ODF) for Canada. J Popul Ther Clin Pharmacol. 2014;21(1):e42-e46.
- Minister Ambrose announces patient involvement pilot for orphan drugs. News Release [Internet]. Ottawa: Government of Canada; 2014 Aug 6. [cited 2015 Sep 22]. Available from: http://news.gc.ca/web/article-en.do?nid=873619
- Hadjivasiliou A. Orphan drug report 2014 [Internet]. London: EvaluatePharma; 2014. [cited 2015 Sep 23]. Available from: http://info.evaluategroup.com/rs/evaluatepharmaltd/images/2014OD.pdf
- Worldwide orphan drug sales and share of prescription drug market (2000 to 2020) [Internet]. In: Orphan drug report. EvaluatePharma; 2014. p. 7 [cited 2015 Sep 30]. Available from: http://info.evaluategroup.com/rs/evaluatepharmaltd/images/2014OD.pdf.
- Pharmaceutical Research and Manufacturers of America (PhRMA). PhRMApedia press room [Internet]. Washington(DC). Record number of medicines in development for rare diseases [news release]; 2011 Feb 24 [cited 2015 Dec 4]. Available from: http://www.phrma.org/media/releases/record-number-medicines-development-rare-diseases
- Rare diseases: a report on orphan drugs in the pipeline: 2013 report [Internet]. Washington (DC): Pharmaceutical Research and Manufacturers of America; 2013. [cited 2015 Sep 17]. Available from: http://www.phrma.org/sites/default/files/pdf/Rare_Diseases_2013.pdf
- Short H, Stafinski T, Menon D. A national approach to reimbursement decision-making on drugs for rare diseases in Canada? Insights from across the ponds. Healthcare Policy. 2015 May;10(4):24-46.
- Pharmaceutical Benefits Advisory Committee. Guidelines for preparing submissions to the pharmaceutical benefits advisory committee [Internet]. Version 4.4. Canberra, Australia: Australian Government, Department of Health; 2013 Jun. [cited 2015 Sep 22]. Available from: http://www.pbac.pbs.gov.au/content/information/printable-files/pbacg-book.pdf
- Department of Health and Ageing [Internet]. Canberra: The Commonwealth of Australia, Department of Health and Ageing; 2013. Life saving drugs program criteria and conditions; 2013 Apr 29 [cited 2015 Nov 26]. Available from: http://www.health.gov.au/internet/main/publishing.nsf/Content/lsdp-criteria
- Australian Government. Department of Health [Internet]. The Department. Other supply arrangements outside the Pharmaceutical Benefits Scheme (PBS); 2015 Oct 1 [cited 2015 Dec 18]. Available from: http://www.health.gov.au/lsdp
- Kanavos P, Nicod E, van den Aardweg S, Pomedli S. The impact of health technology assessments: an international comparison. Euro Observer [Internet]. 2010 [cited 2015 Nov 26];12(4):1-7. Available from: http://www.euro.who.int/__data/assets/pdf_file/0007/127798/EuroObserver_Vol12_No4_Winter_-2010.pdf
- Gemeinsamer Bundesausschuss. Benefits Assessment of Pharmaceuticals (§35a SGB V) [Internet]. The Committee. Questions and answers about the procedure [date unknown]; [cited 2015 Dec 18]. Available from: http://www.english.g-ba.de/benefitassessment/information/faq/#3
- PACE. Medicines for end of life and very rare conditions [Internet]. Glasgow: Scottish Medicines Consortium; 2014 Apr. [cited 2015 Sep 22]. Available from: http://www.scottishmedicines.org/Submission_Process/Submission_guidance_and_forms/PACE
- PACE (patient & clinician engagement) overview document. Process changes for end of life and very rare conditions (orphan and ultra-orphan medicines) [Internet]. Glasgow: Scottish Medicines Consortium; 2014 Apr. [cited 2016 Jan 28]. Available from: http://www.scottishmedicines.org/files/PACE/PACE_Overview_Document_FINAL.pdf
- Sculpher M. Cost-effectiveness evidence to inform drug reimbursement decisions: Are countries converging in process and methods? [Internet]. York: University of York; 2010 Sep. [cited 2015 Nov 26]. Available from: http://www.york.ac.uk/media/che/documents/papers/presentations/Madrid%20talk%20Sept%2010.pdf
- NICE highly specialised technologies guidance [Internet]. London: National Institute for Health and Care Excellence; 2014. [cited 2015 Sep 22]. Available from: http://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-highly-specialised-technologies-guidance
- Interim process and methods of the highly specialised technologies programme [Internet]. London: National Institute for Health and Care Excellence; 2013 May. [cited 2015 Sep 22]. Available from: http://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-highly-specialised-technologies-guidance/Highly-Specialised-Technologies-Interim-methods-and-process-statements.pdf
- Menon D, Clark D, Stafinski T. Reimbursement of drugs for rare diseases through the public healthcare system in Canada: Where are we now? Healthc Policy. 2015 Aug;11(1):15-32.
- New Brunswick drugs for rare diseases plan [Internet]. Fredricton (NB): Government of New Brunswick; 2015. [cited 2015 Sep 22]. Available from: http://www2.gnb.ca/content/gnb/en/services/services_renderer.201352.New_Brunswick_Drugs_for_Rare_Diseases_Plan.html
- Alberta Health and Wellness. Alberta rare diseases drug program: fact sheet [Internet]. Edmonton: The Ministry; 2008. [cited 2015 Nov 26]. Available from: http://www.health.alberta.ca/documents/Pharma-Strategy-2008-rare-disease.pdf
- Alberta Blue Cross, Pharmacy Services. Rare diseases drug coverage program [Internet]. In: Edmonton: Alberta Blue Cross; 2015 Apr 1. Chapter 4 [cited 2015 Nov 26]. Available from: https://www.ab.bluecross.ca/dbl/pdfs/dbl_sec4.pdf.
- CTV. CTV news [Internet]. Calgary. New program for pricey drugs; 2012 Mar 2 [cited 2015 Nov 26]. Available from: http://calgary.ctvnews.ca/new-program-for-pricey-drugs-1.776050
- Winquist E, Bell CM, Clarke JT, Evans G, Martin J, Sabharwal M, et al. An evaluation framework for funding drugs for rare diseases. Value Health [Internet]. 2012 [cited 2015 Nov 26];15(6):982-6. Available from: http://www.sciencedirect.com/science/article/pii/S1098301512016221
- Chan W. Drugs for rare diseases: Ontario experience: Canada's Drug Agency teleconference. Ottawa: Canadian Agency for Drugs and Technologies in Health; 2013 Jan 15.
- Winquist E, Coyle D, Clarke JT, Evans GA, Seager C, Chan W, et al. Application of a policy framework for the public funding of drugs for rare diseases. J Gen Intern Med [Internet]. 2014 Aug [cited 2015 Nov 26];29 Suppl 3:S774-9, 2014 Aug.:-9. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4124122
- Food and Drug Administration OoOPD. Drugs [Internet]. Silver Spring (MD): FDA. For industry: Developing products for rare diseases & conditions; 2015 Oct 19 [cited 2015 Nov 26]. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
- U.S. Food and Drug Administration. Verification of orphan-drug status. Subpart 316.21. 2015 Nov 24 [cited 2016 Jan 26]. In: U.S. Government Printing Office. e-CFR.gov [Internet]. Washington(DC). Available from: http://www.ecfr.gov/cgi-bin/text-idx?c=ecfr&SID=a731e05779ee0cae2c87b21a8739eae9&rgn=div8&view=text&node=21:5.0.1.1.6.3.1.2&idno=21.
- U.S.Food and Drug Administration. Orphan Drug Act [excerpts]: Congresssional findings for the Orphan Drug Act [Internet]. amended. In: Federal Food, Drug, and Cosmetic Act (FD&C Act). Silver Spring(MD): FDA; 2013 Jun 12 [cited 2013 Aug 6]. Available from: http://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=0e737d105ef9a1632b19a1e713b93cc4&mc=true&n=pt21.5.316&r=PART&ty=HTML.
- U.S. Food and Drug Administration. Designating an orphan product: drugs and biologics [Internet]. Silver Spring ( MD): FDA. 21 CFR PART 316 and Final Rule December 29, 1992 [orphan drugs]; 2015 Nov 24 [cited 2015 Nov 26]. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/ucm124562.htm
- Seoane-Vazquez E, Rodriguez-Monguio R, Szeinbach SL, Visaria J. Incentives for orphan drug research and development in the United States. Orphanet J Rare Dis [Internet]. 2008 [cited 2015 Nov 26];3:33. Available from: http://www.ojrd.com/content/pdf/1750-1172-3-33.pdf
- CDER Small Business Assistance. Orphan drugs [Internet]. Silver Spring (MD): Food and Drug Administration, Office of Communication, Division of Drug Information; 2012 Jul 13. [cited 2015 Sep 22]. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/UCM311928.pdf
- European Commission. COMMISSION REGULATION (EC) No 847/2000 of 27 April 2000 laying down the provisions for implementation of the criteria for designation of a medicinal
product as an orphan medicinal product and definitions of the concepts 'similar medicinal product' and 'clinical superiority'. 2000 Apr 28 [cited 2015 Nov 26]. In: EU Publications Office. EUR-LEX.europa.eu [Internet]. EUR-Lex. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2000:103:0005:0008:EN:PDF. - European Medicines Agency. Marketing authorisation and market exclusivity [Internet]. London: European Medicines Agency; [cited 2015 Sep 22]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000392.jsp&mid=WC0b01ac058061f019
- European Medicines Agency. Orphan incentives [Internet]. London: The Agency; [cited 2015 Sep 22]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000393.jsp&mid=WC0b01ac0580024c5a
- European Parliament. Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. 2000 [cited 2015 Nov 26]. In: EU Publications Office. EUR-LEX.europa.eu [Internet]. Brussels: EUR-Lex. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2000:018:0001:0005:en:PDF.
- Gammie T, Lu CY, Babar ZU. Access to orphan drugs: A comprehensive review of legislations, regulations and policies in 35 countries. PLoS One [Internet]. 2015 [cited 2015 Oct 28];10(10):e0140002, 2015. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4599885/pdf/pone.0140002.pdf
- Johnson JA, Thaul S, Bagalman E. H.R. 6: The 21st Century Cures Act. Washington (DC): Library of Congress - Congressional Research Service; 2015 Aug 10. [cited 2016 Jan 25].
- Fast track [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2014 Sep 14. [cited 2016 Jan 25]. Available from: http://www.fda.gov/ForPatients/Approvals/Fast/ucm405399.htm
- Breakthrough therapy [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2014 Sep 15. [cited 2016 Jan 25]. Available from: http://www.fda.gov/ForPatients/Approvals/Fast/ucm405397.htm
- Health Canada. [Internet]. Ottawa: Government of Canada. Guidance for industry - Priority review of drug submissions; 2012 Jul 6 [cited 2016 Jan 25]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/priorit/priordr-eng.php#a1.2
- Priority review [Internet]. Silver Spring (MD): U.S. Department of Health & Human Services, U.S. Food and Drug Administration; 2014 Sep 15. [cited 2016 Jan 25]. Available from: http://www.fda.gov/ForPatients/Approvals/Fast/ucm405405.htm
About this Document
Author: Lili Loorand-Stiver, Tara Cowling, Christine Perras.
Disclaimer: The Canadian Agency for Drugs and Technologies in Health (CADTH) takes sole responsibility for the final form and content of this environmental scan. The statements and conclusions in this environmental scan are those of Canada's Drug Agency. The Environmental Scanning Service is an information service for those involved in planning and providing health care in Canada. Environmental Scanning Service responses are based on a limited literature search and are not comprehensive, systematic reviews. The intent is to provide information on a topic that Canada's Drug Agency could identify using all reasonable efforts within the time allowed. Environmental Scanning Service responses should be considered along with other types of information and health care considerations. The information included in this response is not intended to replace professional medical advice nor should it be construed as a recommendation for or against the use of a particular health technology. Readers are also cautioned that a lack of good quality evidence does not necessarily mean a lack of effectiveness, particularly in the case of new and emerging health technologies for which little information can be found but that may in future prove to be effective. While Canada's Drug Agency has taken care in the preparation of the report to ensure that its contents are accurate, complete, and up to date, Canada's Drug Agency does not make any guarantee to that effect. Canada's Drug Agency is not liable for any loss or damages resulting from use of the information in the report. Links: This report may contain links to other information available on the websites of third parties on the Internet. Canada's Drug Agency does not have control over the content of such sites. Use of third party sites is governed by the owners’ own terms and conditions.
Copyright© Canada's Drug Agency 2016. You are permitted to make copies of this document for non-commercial purposes provided it is not modified when reproduced and appropriate credit is given to Canada's Drug Agency.
About Canada's Drug Agency: Canada's Drug Agency is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: Canada's Drug Agency receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
Views: The views expressed herein are those of Canada's Drug Agency and do not necessarily reflect the views of our funders.
Cite as: Drugs for rare diseases: evolving trends in regulatory and health technology assessment perspectives. Ottawa; Canada's Drug Agency; 2013 Oct [updated 2016 Feb]. (Environmental scan; issue 42).
Contact [email protected] with inquiries about this notice or legal matters relating to Canada's Drug Agency services.
Files
Last Updated : February 11, 2016